Warning
You require the optional dependency SciPy package to use this script. If you are unsure if it is installed on your system, refer to the Installation documentation
1. The MSA ConKit hierarchy needs to be created first.
>>> import conkit.io
>>> msa = conkit.io.read('toxd/toxd.a3m', 'a3m')
2. To obtain the length of the target sequence, you can simply ask the “msa“ hierarchy for it.
>>> print('Length of the Target Sequence: %d' % msa.top_sequence.seq_len)
59
This tells you the first sequence in the alignment has 59 residues, i.e. the chain length of your target.
3. We can again use the “msa“ hierarchy to figure out the total number of sequences.
>>> print('Total number of sequences: %d' % msa.nseq)
Total number of sequences: 13488
4. … and the number of effective sequences in the alignment at 70% identity cutoff.
>>> n_eff = msa.neff
>>> print('Number of Effective Sequences: %d' % n_eff)
Number of Effective Sequences: 3318
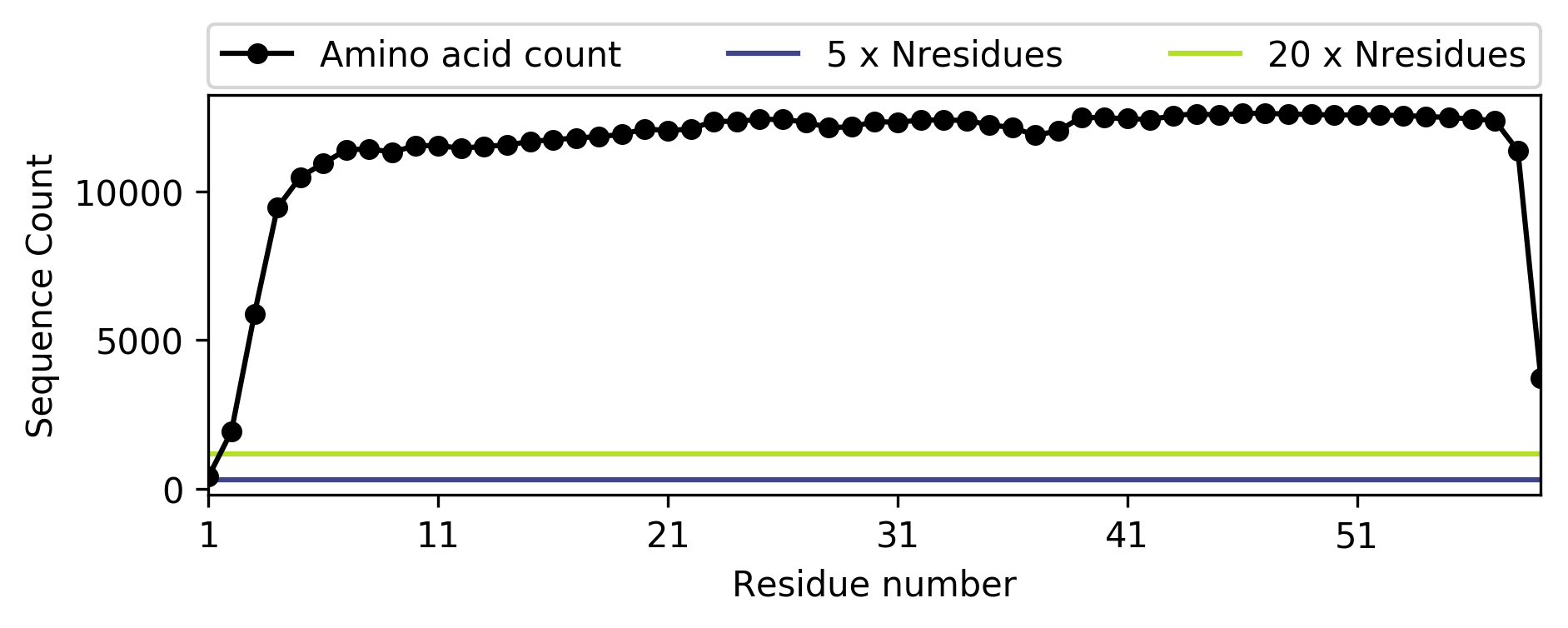
5. We can also plot the amino acid frequency at each position in the alignment.
>>> file_name = 'toxd/toxd.png'
>>> import conkit.plot
>>> conkit.plot.SequenceCoverageFigure(msa, file_name=file_name)